Infiltrative cardiomyopathies are a diverse group of cardiac diseases caused by the deposition of abnormal substances within the myocardium and include diseases, such as Fabry disease, sarcoidosis and less common genetic diseases, such as Friedreich’s ataxia and Danon disease.1 Cardiac amyloidosis is the exemplar infiltrative cardiomyopathy and is characterized by the deposition of misfolded precursor proteins into insoluble, protease-resistant amyloid fibrils, which accumulate in the myocardial extracellular matrix.2 The deposition of amyloid fibrils disrupt the cardiac structure and function, typically resulting in biventricular wall thickening, stiffening of the myocardium and the development of restrictive physiology and systolic dysfunction.2 Over 30 different human precursor proteins can form amyloid fibrils, but the majority of cardiac amyloidosis cases result from misfolded transthyretin (transthyretin cardiac amyloidosis [ATTR-CA]) and immunoglobulin light-chain (light-chain amyloidosis [AL]) proteins. Less common, although increasingly recognized, causes of cardiac amyloidosis include apolipoprotein AI amyloidosis and apolipoprotein AIV amyloidosis.3–5
ATTR-CA represents an important cause of heart failure among older individuals.6 In the sporadic, non-inherited, wild-type form, misfolding occurs secondary to a pathological process that is associated with age-related homeostatic mechanisms; this is a condition of older, predominantly male individuals. However, the hereditary form occurs secondary to a single-point mutation in the transthyretin (TTR) gene and often presents with a varying clinical phenotype, often comprising both cardiomyopathy and a length-dependent sensorimotor peripheral polyneuropathy and/or autonomic neuropathy.6–8
Advances in cardiac imaging, combined with increased awareness among clinicians, have resulted in significantly increased diagnoses in recent years. What was once thought of as a rare disease is increasingly recognized as an important cause of heart failure, especially in older individuals.9 Furthermore, the variant most commonly associated with ATTR-CA (val122Ile) is present in 3–4% of African Americans, with an estimated 1.5 million individuals in the USA being allele carriers.10
Until recently, the mainstay of management for patients with ATTR-CA was supportive therapy using loop diuretics to aid meticulous volume control and treatment of comorbidities, such as anticoagulation in the presence of atrial fibrillation.11,12 A recent study of heart failure medications in patients with ATTR-CA demonstrated that mineralocorticoid receptor antagonists and low-dose beta-blockers in patients with a reduced ejection fraction were associated with improved survival; however, neither of these conventional heart failure medications target the specific pathways responsible for ATTR amyloid fibril formation.13 A deeper understanding of the underlying pathophysiology has resulted in the discovery of multiple, different, disease-specific pharmacotherapies that are either approved for clinical use or at different stages of development.14 This review will explore the current therapeutic strategies available for patients with ATTR-CA and provide insights into future perspectives.
Transthyretin stabilizers
Current therapeutic strategies are aimed at reducing the formation of ATTR amyloid fibrils and the subsequent deposition within the myocardium to slow disease progression.15,16 The discovery of the Thr119Met TTR gene polymorphism, which encodes an amino acid substitution that stabilizes the transthyretin protein, even in the context of known destabilizing pathogenic TTR variants, spurred the development of transthyretin stabilizers. These agents bind to the transthyretin tetramer to prevent the dissociation into amyloidogenic monomers and oligomers that subsequently form pathogenic amyloid fibrils.15,16
Diflunisal
Diflunisal is a non-steroidal anti-inflammatory drug, which binds to the two thyroxine-binding sites on the transthyretin protein and kinetically stabilizes the tetramer to prevent the dissociation into amyloidogenic monomers.17 Diflunisal was initially studied in patients with ATTR-polyneuropathy (ATTR-PN), and in a phase III placebo-controlled trial it was shown to reduce the progression of neurological symptoms.17
Experience with diflunisal in patients with ATTR-CA has been limited to small open-label studies. Imaging-based studies suggest that treatment with diflunisal may slow the progression of echocardiographic decline, with small studies demonstrating the stabilization of global longitudinal strain (GLS) measurements.18,19 A retrospective analysis of 120 patients (13 treated with diflunisal, 16 treated with tafamidis and 91 not treated with a TTR stabilizer) demonstrated that treatment with TTR stabilizers was associated with a reduction in mortality and orthoptic heart transplant, and diflunisal was associated with a similar survival benefit to tafamidis.20 A further retrospective analysis of 104 patients with ATTR-CA, 35 of whom were treated with diflunisal, concluded that treatment with diflunisal was associated with improved survival. However, it is noteworthy that the treated and untreated patients were poorly matched, with the treated patients being younger and having a milder cardiac phenotype.21
Due to the lack of large-scale clinical trials, diflunisal remains unlicensed for the treatment of ATTR-CA, and it should be used with care as a non-steroidal anti-inflammatory drug, avoided in patients with renal impairment and a history of previous gastrointestinal bleeding, and used with caution in patients with recent heart failure decompensation or requiring high-dose diuretics.22
Tafamidis
Tafamidis is a benzoxazole derivative lacking non-steroidal anti-inflammatory properties, which binds with high affinity and selectivity to the transthyretin thyroxine-binding sites, inhibiting dissociation. A small phase II trial (The Effects Of Fx-1006A On Transthyretin Stabilization And Clinical Outcome Measures In Patients With V122I Or Wild-Type TTR Amyloid Cardiomyopathy; ClinicalTrials.gov identifier: NCT00694161) of 31 patients with ATTR-CA demonstrated that tafamidis stabilized transthyretin with an acceptable safety profile.23 The ATTR-ACT trial (A Multicenter, International, Phase 3, Double-Blind, Placebo-Controlled, Randomized Study To Evaluate The Efficacy, Safety, And Tolerability Of Daily Oral Dosing Of Tafamidis Meglumine (PF-06291826) 20 mg Or 80 mg In Comparison to Placebo in Subjects Diagnosed With Transthyretin Cardiomyopathy (TTR-CM); ClinicalTrials.gov identifier: NCT01994889) was a phase III, randomized, placebo-controlled double-blind trial of 441 patients with ATTR-CA, which assessed the efficacy of tafamidis.24 The primary analysis used the Finkelstein–Schoenfeld method, which is a hierarchical rank-sum analysis where all-cause mortality was assessed first, followed by cardiovascular hospitalizations. The primary endpoint was achieved with a significant win ratio; this was supported by a more traditional time to the first event analysis where tafamidis reduced both all-cause mortality and heart failure hospitalizations. Treatment with tafamidis also resulted in a slower decline in the 6-minute walk test distance and the Kansas City Cardiomyopathy Questionnaire (KCCQ) score compared with placebo.24 A post-hoc analysis of 436 patients with available echocardiographic data demonstrated that treatment with tafamidis was associated with a less pronounced worsening of stroke volume, GLS and diastolic function, as measured by E/e’.25 Following the ATTR-ACT trial, tafamidis was subsequently awarded breakthrough designation by the US Food and Drug Association (FDA) for the treatment of ATTR-CA. At present it is the only disease-modifying therapy currently licenced for this indication.22,26
The efficacy of long-term use of tafamidis was explored in the long-term extension study, whereby all patients enrolled on the ATTR-ACT trial either continued tafamidis or, if randomized to placebo, were initiated on tafamidis.27 Mortality was reduced in patients treated with continuous tafamidis compared with those who were switched from placebo to tafamidis, indicating the importance of early initiation of disease-modifying treatment.
Acoramidis
Acoramidis is a novel TTR stabilizer designed to mimic the action of the Thr119Met variant and achieves near-complete TTR stabilization.28 When compared with diflunisal and tafamidis, acoramidis has shown improved binding affinity, binding thermodynamics, binding-site occupancy and stabilization.28–30 Improved stabilization has been attributed to acoramidis using a primarily enthalpic binding mode, which mimics the hydrogen bonding seen in the Thr119Met variant, as compared with the entropic binding mode of diflunisal and tafamidis.28–30
The ATTRibute-CM trial (A Phase 3, Randomized, Double-Blind, Placebo-Controlled Study of the Efficacy and Safety of AG10 in Subjects With Symptomatic Transthyretin Amyloid Cardiomyopathy [ATTRibute-CM Trial; ClinicalTrials.gov identifier: NCT03860935) was a phase III, randomized, controlled double-blind trial of 632 patients with ATTR-CA, which assessed the efficacy of acoramidis.31 The analysis used the Finkelstein–Schoenfeld method (similar to the ATTR-ACT trial) and used a four-step hierarchical analysis that included all-cause mortality, the cumulative frequency of cardiovascular-related hospitalization, the change from baseline in the N-terminal pro-B-type natriuretic peptide (NT-proBNP) level and the change from baseline in the 6-minute walk test distance. The primary endpoint was achieved with a significant win ratio, and the prespecified secondary analyses, performed according to either the two-component hierarchy of death and cardiovascular-related hospitalization or the three-component hierarchy with the addition of a change in the 6-minute walk test distance, also both yielded significant win ratios. Treatment with acoramidis also resulted in a slower rate of decline in the KCCQ score.31 The US FDA has accepted a new drug application for acoramidis for the treatment of ATTR-CA, and this application is due to be reviewed in the near future.32
Gene silencers
Prior to the development of specific disease-modifying therapies, there were reports of patients with ATTR-PN being successfully treated with liver transplantation, which suppressed the production of the variant TTR and prevented the further accumulation of amyloid fibrils within the nerves.33 Successful outcomes supported the hypothesis that reducing or halting TTR production could prevent disease progression and led to the development of medicines that target hepatic TTR synthesis through the disruption of the relevant messenger RNA (mRNA).34
Patisiran
Patisiran is a first-generation small-interfering RNA (siRNA) molecule formulated in a lipid nanoparticle to ensure targeted delivery to hepatocytes.35 Upon entry into the cytoplasm, the double-stranded siRNA splits into single-stranded RNA, which bind mRNA coding for the TTR protein. The binding process activates the Argonaute slicer protein, which degrades mRNA, and inhibits TTR synthesis.36,37 Patisiran has already demonstrated efficacy in patients with ATTR-PN. The APOLLO trial (APOLLO: A Phase 3 Multicenter, Multinational, Randomized, Double-blind, Placebo-controlled Study to Evaluate the Efficacy and Safety of Patisiran [ALN-TTR02] in Transthyretin [TTR]-Mediated Polyneuropathy [Familial Amyloidotic Polyneuropathy-FAP]; ClinicalTrials.gov identifier: NCT01960348) randomized 225 patients with ATTR-PN to patisiran or placebo and demonstrated that patisiran treatment improved neurological outcomes.38 A post-hoc analysis evaluated the impact of patisiran on cardiac outcomes in 126 patients from the APOLLO trial who were deemed to have cardiac amyloidosis based on the prespecified criteria of a left ventricular wall thickness of ≥13 mm in the absence of aortic valve disease or hypertension.39 Patisiran-treated patients had reduced left ventricular wall thickening, increased left ventricular end-diastolic volume and more favourable changes in GLS compared with those on placebo. The greatest differential improvement in strain was observed in the basal region, suggesting that basal longitudinal strain may be a more sensitive marker of treatment associations.40 The treated group also had a significant reduction in NT-proBNP and an increase in 10-meter walk test gait speed compared with the placebo group. However, the definition of cardiac involvement is non-specific and therefore could have resulted in the inclusion of patients who do not fulfil the guideline-mandated diagnostic criteria for ATTR-CA.39
These findings were supported by a study of 16 patients treated with patisiran and diflunisal, which demonstrated a reduction in cardiac magnetic resonance-derived extracellular volume, serum TTR and NT-proBNP, and an increase in the 6-minute walk test distance and a reduction in cardiac uptake on bone scintigraphy following 12 months of treatment.41 Although the reduced cardiac uptake unequivocally indicated a favourable effect of patisiran, the kinetics and dynamics of radiotracer binding to bones, soft tissues and the myocardium differ, and a change in any compartment will affect the calculation of proportionate cardiac uptake. Therefore, reduced cardiac uptake should be supported by improvements in other cardiac parameters before being ascribed exclusively to a decrease in amyloid burden.41
Following the successful outcomes in patients with ATTR-PN, the efficacy of patisiran for the treatment of ATTR-CA was evaluated in the APOLLO-B trial (APOLLO-B: A Phase 3, Randomized, Double-blind, Placebo-controlled Multicenter Study to Evaluate the Efficacy and Safety of Patisiran in Patients With Transthyretin Amyloidosis With Cardiomyopathy [ATTR Amyloidosis With Cardiomyopathy]; ClinicalTrials.gov identifier: NCT03997383).42 The APOLLO-B trial was a phase III, double-blind, randomized controlled trial of 360 patients with ATTR-CA. It met its primary endpoint with a slower decline in the 6-minute walk test distance among patients treated with patisiran compared with placebo at 1 year, with a mean difference of 14.7 m. This was accompanied by a slight increase in KCCQ scores in the patisiran group, while KCCQ scores decreased in the placebo group. Exploratory endpoints demonstrated that patisiran treatment also slowed the increase in NT-proBNP and troponin-I compared with placebo. However, despite the treatment with patisiran slowing the decline in the 6-minute walk test distance, the FDA declined the application to approve patisiran for the treatment of ATTR-CA, with the panel expressing concerns over the trial design and the effect size being small and of questionable meaningfulness.43
Vutrisian
Vutrisian is a second-generation siRNA that uses a N-acetylgalactosamine (GalNAc) conjugate delivery platform, which enhances stabilization chemistry and allows subcutaneous administration of smaller doses with longer dosing intervals.44 Therefore, vutrisian can be administered as a subcutaneous injection every 3 months, rather than the 3-weekly intravenous infusion required to administer patisiran.44 Vutrisian has already demonstrated efficacy in patients with ATTR-PN.45 The HELIOS-A trial (HELIOS-A: A Phase 3 Global, Randomized, Open-label Study to Evaluate the Efficacy and Safety of ALN-TTRSC02 in Patients With Hereditary Transthyretin Amyloidosis [hATTR Amyloidosis]; ClinicalTrialsgov identifier: NCT03759379) randomized 164 patients with ATTR-PN to vutrisiran or patisiran and compared the outcomes with an external placebo group from the APOLLO trial. The trial demonstrated that vutrisiran treatment was associated with improved neurological outcomes.45 A post-hoc exploratory analysis evaluated the impact of vutrisiran on cardiac outcomes by comparing the outcomes in patients treated with vutrisiran in the HELIOS-A trial with a historical placebo group from the APOLLO trial. A subgroup analysis of patients considered to have cardiac amyloidosis based on having a left ventricular wall thickness of ≥13 mm in the absence of aortic valve disease or hypertension was also carried out. In the overall study population, and those considered to have cardiac involvement, treatment with vutrisiran was associated with beneficial changes in NT-proBNP compared with placebo. Vutrisiran was also associated with a nominally significant benefit in cardiac output and stroke volume compared with placebo in the overall population, and the subgroup considered to have cardiac involvement. It is noteworthy that this study used a historical placebo group from the APOLLO trial, and there were significant differences in baseline characteristics between the treatment and placebo groups. Furthermore, similar to the post-hoc analysis of the APOLLO trial, the definition of cardiac involvement is non-specific and therefore could have resulted in the inclusion of patients who do not fulfil the guideline-mandated diagnostic criteria for ATTR-CA.46
The efficacy of vutrisian treatment in patients with ATTR-CA is currently being assessed in the HELIOS-B trial (HELIOS-B: A Phase 3, Randomized, Double-blind, Placebo-controlled, Multicenter Study to Evaluate the Efficacy and Safety of Vutrisiran in Patients With Transthyretin Amyloidosis With Cardiomyopathy [ATTR Amyloidosis With Cardiomyopathy]; ClinicalTrials.gov identifier: NCT04153149), is a randomized, placebo-controlled, double-blind trial of 665 patients with ATTR-CA.47 The primary endpoint is a composite of all-cause mortality, cardiovascular hospitalizations and urgent heart failure hospital visits. The secondary endpoints include changes from the baseline in the 6-minute walk test distance, the KCCQ score, NT-proBNP, mean left ventricular wall thickness and GLS.
Inotersen
Inotersen is a first-generation antisense oligonucleotide (ASO).48–50 ASOs typically consist of a single strand of 16–20 synthetic nucleotides, which bind to their complementary mRNA and induce degradation. Inotersen is a 2’-O-methoxyethyl-modified ASO, which binds to the 3’-untranslated portion of the complementary mRNA and promotes the mRNA breakdown by activating the ribonuclease H1-mediated degradation pathway.48–50 The NEURO-TTR trial (A Phase 2/3 Randomized, Double-Blind, Placebo-Controlled Study to Assess the Efficacy and Safety of ISIS 420915 in Patients With Familial Amyloid Polyneuropathy [NEURO-TTR Study]; ClinicalTrials.gov identifier: NCT01737398) was a phase III, randomized, placebo-controlled double-blind study that demonstrated the efficacy of inotersen in treating patients with ATTR-PN.51 However, the change in echocardiographic parameters, including GLS, did not differ significantly between the treatment and placebo groups. The assessment of inotersen in patients with ATTR-CA is limited to small imaging-based studies. A small study of 30 patients with ATTR-CA demonstrated that inotersen stabilized cardiac disease across a range of echocardiographic and cardiac magnetic resonance parameters after 12 months of treatment.52 Another study of 33 patients demonstrated that treatment with inotersen was associated with an increase in the 6-minute walk test distance and a reduction in left ventricular mass (as measured by cardiac magnetic resonance) at both 24 and 36 months.53 The efficacy of inotersen is also being assessed in an open-label phase II study of 50 patients with ATTR-CA.54 This study will assess the change in echocardiographic GLS and cardiac magnetic resonance parameters (left ventricular mass and extracellular volume) at 6, 12 and 24 months. The efficacy of inotersen in patients with ATTR-CA is yet to be assessed in a large phase III trial, and therefore, it has only been approved for the treatment of ATTR-PN.55
Eplontersen
Eplontersen is a second-generation ASO, with a similar design to inotersen, but is conjugated to a triantennary GalNAc moiety, which facilitates the receptor-mediated uptake into the target hepatocytes.56,57 This superior delivery system has enabled eplontersen to be up to 50-fold more potent than inotersen at reducing TTR expression, which in turn allows it to be administered every 4 weeks, rather than the weekly subcutaneous injection required to administer inotersen.58 The NEURO-TTRansform trial (A Phase 3 Global, Open-Label, Randomized Study to Evaluate the Efficacy and Safety of ION-682884 in Patients With Hereditary Transthyretin-Mediated Amyloid Polyneuropathy; ClinicalTrials.gov identifier: NCT04136184) was a phase III, multicentre randomized trial, designed to assess the efficacy and safety of eplontersen in the treatment of ATTR-PN.59 The trial randomized patients to receive eplontersen or inotersen, and the placebo group from the NEURO-TTR trial served as a historical control. Treatment with eplontersen resulted in improved neurological outcomes compared with placebo. A post-hoc exploratory analysis of the change in echocardiographic parameters in patients treated with eplontersen compared with the historical placebo group demonstrated that in the overall population, eplontersen was associated with improvements in left ventricular end-diastolic volume, stroke volume, E/e’ and left atrial volume. In a subgroup of patients considered to have cardiac amyloidosis based on having a left ventricular wall thickness of ≥13 mm in the absence of aortic valve disease or hypertension, treatment with eplontersen was associated with an improvement in left ventricular stroke volume and left ventricular ejection fraction. However, due to the study design and utilization of a historical cohort from the NEURO-TTR trial, this study is fraught with the same limitations as the post-hoc analysis of patients treated with vutrisiran.60
The CARDIO-TTRansform trial (A Phase 3 Global, Double-Blind, Randomized, Placebo-Controlled Study to Evaluate the Efficacy and Safety of ION-682884 in Patients With Transthyretin-Mediated Amyloid Cardiomyopathy [ATTR CM]; ClinicalTrials.gov identifier: NCT04136171) is a phase III, double-blind, randomized placebo-controlled trial, designed to assess the efficacy of eplontersen in ATTR-CA.61 The trial has completed the enrolment of 1,400 patients and will assess a primary endpoint of a composite of cardiovascular mortality and recurrent cardiovascular events. Key secondary endpoints include a change in the 6-minute walk test distance and the KCCQ score.
Gene-editing therapy
Gene silencing-based therapies that use siRNA and the ASO technology to induce TTR knockdown are clinically beneficial in patients with ATTR amyloidosis; however, these treatments require repeated doses to maintain a therapeutic effect.34 Gene-editing therapy potentially offers a single-dose treatment for ATTR amyloidosis.34 NTLA-2001 has harnessed the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9 technology to edit the gene coding for TTR in hepatocytes.34 CRISPR sequences are transcribed into small RNA capable of guiding the system to complementary DNA sequences.62,63 The Cas9 endonuclease subsequently binds the DNA and cleaves it, knocking out the target gene.64 The circulating TTR protein is exclusively synthesized by the liver and coded for by a single gene, making ATTR amyloidosis the prototypic disease model for targeted in-vivo genome-editing therapy.65
Following the successful administration of NTLA-2001 in various animal models, which consistently resulted in a durable TTR knockdown, NTLA-2001 was tested in a small cohort of patients with ATTR amyloidosis.66 The phase I study of six patients with either ATTR-CA or ATTR-PN demonstrated that a single dose of NTLA-2001 resulted in a sustained TTR knockdown and was well tolerated without any reports of serious adverse events.66 The efficacy of NTLA-2001 in patients with ATTR-CA will be assessed in a phase III, double-blind, randomized placebo-controlled trial, which will enrol 765 patients and assess the impact of NTLA-2001 on the primary endpoint of a composite of cardiovascular mortality and cardiovascular events.67 Key secondary endpoints include the change in the KCCQ score and serum TTR levels.
Anti-amyloid therapies
Through various different mechanisms, the aforementioned therapies all reduce the formation of ATTR amyloid fibrils, but do not facilitate the removal of amyloid fibrils that have already accumulated in the myocardium.34 Amyloid removal represents an important unmet clinical need, especially considering that many patients still present with advanced cardiac disease, and can rapidly progress to end-stage heart failure with multiorgan dysfunction and recurrent hospitalizations.68–70
The concept of amyloid removal and disease regression has been more widely characterized in other forms of systemic amyloidosis, such as in systemic AA amyloidosis and systemic AL amyloidosis, whereby a deep knockdown of the amyloid precursor protein (serum amyloid A protein in systemic AA amyloidosis and serum free light chain in systemic AL amyloidosis) can result in a reduction of the amyloid burden.38,71,72 This was first described in the visceral organs using serial serum amyloid P component scintigraphy scans (a qualitative nuclear medicine technique used to visualize the visceral amyloid burden), and more recently has been described in the heart using cardiac magnetic resonance imaging with multiparametric mapping, with amyloid removal being associated with favourable biochemical, structural and functional changes.73–76
Amyloid accumulation is dynamic and involves constant turnover, whereby disease progression occurs if the rate of formation exceeds the rate of intrinsic clearance, and disease regression occurs if the rate of intrinsic clearance exceeds the rate of formation.77 However, the natural clearance of amyloid fibrils is a slow and arduous process that could take years.73,78 Therefore, therapies that accelerate amyloid fibril removal would represent a significant advancement in the treatment of cardiac amyloidosis.
The antibody-mediated removal of ATTR amyloid fibrils was recently described in three patients who spontaneously developed polyclonal immunoglobulin G (IgG) antibodies to human ATTR amyloid, which was associated with reversion to near-normal cardiac structure and function, in the absence of any disease-modifying therapy.79 Clinical recovery was corroborated by substantial improvements in symptom burden, functional capacity, serum biochemistry and across a range of cardiac imaging parameters. Cardiac magnetic resonance with multiparametric mapping confirmed near-complete disease regression, without residual scarring. This exciting discovery has provided proof of concept that amyloid removal can be facilitated through antibody-based therapies and result in disease reversal.79
NI006
There are several investigational anti-amyloid therapies at various stages of development, of which NI006 is at the most advanced stage.79,80 NI006 is a recombinant human anti-ATTR monoclonal IgG1 antibody that binds ATTR amyloid confirmations of TTR and induces antibody-mediated phagocytosis of ATTR fibrils and subsequent removal of deposits from tissues.80 A phase I, randomized double-blind trial of 40 patients with ATTR-CA demonstrated that NI006 was safe and not associated with serious drug-related adverse events.81 At higher doses, both serum NT-proBNP and troponin-I appeared to be reduced, as did the myocardial extracellular volume, which represents a surrogate marker of cardiac amyloid burden. Cardiac radiotracer uptake on bone scintigraphy also appeared to be reduced, and this reduction was supported by improvements in cardiac biomarkers and extracellular volume, indicating a likely decrease in amyloid burden.81 The efficacy of NI006 in patients with ATTR-CA will be assessed in a phase III, double-blind, randomized placebo-controlled trial (A Phase 3, Randomized, Double-blind, Placebo-controlled, Multicenter Study to Evaluate the Efficacy and Safety of Amyloid Depleter ALXN2220 in Adult Participants With Transthyretin Amyloid Cardiomyopathy [ATTR-CM]; ClinicalTrials.gov identifier: NCT06183931) that will assess the impact of N1006 on the primary endpoint of a composite of all-cause mortality and recurrent cardiovascular events.82 The key secondary endpoints include a change in the 6-minute walk test distance and the KCCQ score.
PRX004
PRX004 is a humanized IgG monoclonal antibody, which targets an epitope that is exposed to misfolded TTR.83 Preclinical models demonstrated that PRX004 was able to neutralize soluble cytotoxic forms of TTR, prevent amyloid fibril formation and additionally promote macrophage-mediated clearance of amyloid fibrils already deposited in tissues.
A phase I dose-escalation study in 21 patients with either ATTR-CA or ATTR-PN demonstrated that PRX004 was safe and well tolerated.84 In all seven evaluable patients, there was an improvement in echocardiographic GLS. It is noteworthy that although these results have been presented, the full manuscript is yet to be published, and therefore, more detail is required before formal conclusions can be drawn.84 A phase II study (Efficacy and Safety of NNC6019-0001 at Two Dose Levels in Participants With Transthyretin Amyloid Cardiomyopathy [ATTR CM]; ClinicalTrials.gov identifier: NCT05442047) has been initiated to assess the use of escalating doses of PRX004 compared with placebo in patients with ATTR-CA.85
AT-02
Perhaps the most exciting anti-amyloid therapy is AT-02, which represents a pan-amyloid removal technology.86 AT-02 is a fusion protein that consists of synthetic peptide linked to a single-chain human immunoglobulin G1 Fc domain. The naturally occurring pentameric serum amyloid P binds to all forms of amyloid with a high affinity. Once bound, the Fc portion of the immunoglobulin engages with the immune system and promotes macrophage-mediated phagocytosis of the amyloid fibrils.86 Therefore, AT-02 has the potential to treat all causes of cardiac amyloidosis, regardless of the underlying form of amyloid. This could represent a significant advancement, as it can sometimes be challenging to confirm the underlying cause of cardiac amyloidosis, with the different types often sharing common features of cardiac imaging.2,4 Preclinical studies have demonstrated that AT-02 binds with a high affinity to ATTR and AL amyloid fibrils and subsequently induces macrophage-mediated phagocytosis.86 A phase II study that will assess the safety of AT-02 in patients with various forms of amyloidosis, including ATTR-CA (A Phase 2, Open-label Extension Study to Evaluate the Long-term Safety and Tolerability of AT-02; ClinicalTrials.gov identifier: NCT05951049), is currently underway (Table 1 and Figure 1).13–16,19,24,30,31,34,36,40–42,44,48,50,57,71,74–76,78,87
Table 1: Summary of therapeutic agents for the treatment of transthyretin cardiac amyloidosis13–16,19,24,30,31,34,36,40–42,44,48,50,57,71,74–76,78
|
Drug |
Mechanism of action |
Stage of approval |
Dose and route of administration |
Cardiovascular trial outcomes |
Side effects |
|
Diflunisal13–16 |
Stabilizes TTR to prevent dissociation |
Approved as an NSAID, off-licence use for ATTR-CA and ATTR-PN |
250 mg orally twice daily |
Small retrospective studies have demonstrated an association with reduced mortality |
Fluid retention, bleeding and renal dysfunction |
|
Tafamidis19 |
Stabilizes TTR to prevent dissociation |
Approved for ATTR-CA and ATTR-PN |
Vyndamax 61 mg orally once daily or Vyndaqel 80 mg orally once daily |
RCT demonstrating a reduction in all-cause mortality and cardiovascular hospitalizations, along with lower rates of the 6MWT and a decline in KCCQ |
None |
|
Acoramidis24 |
Stabilizes TTR to prevent dissociation |
Due to be reviewed by the US FDA |
800 mg orally twice daily |
RCT demonstrating a reduction in the hierarchical composite of all-cause mortality, cardiovascular hospitalizations, change in NT-proBNP and change in 6MWT, along with lower rates of the 6MWT and a decline in KCCQ |
None |
|
Patisiran30,31 |
siRNA targeting TTR mRNA |
Approved for ATTR-PN but not for ATTR-CA |
80 min intravenous infusion based on body weight every 3 weeks: <100 kg = 0.3 mg/kg; >100 kg = 30 mg |
RCT demonstrating a reduction in the rate of the 6MWT decline compared with placebo |
Infusion-related reactions, vitamin A deficiency and peripheral oedema |
|
Vutrisiran34,36 |
siRNA targeting TTR mRNA |
Approved for ATTR-PN but not for ATTR-CA |
25 mg subcutaneous injection every 3 months |
HELIOS-B is a phase III RCT designed to assess the efficacy of vutrisiran in patients with ATTR-CA; the results are likely to be announced in the near future |
Infusion-related reactions, vitamin A deficiency, nasopharyngitis, headache, urinary tract infections and dyslipidaemia |
|
Inotersen40–42,44 |
ASO inhibitor of TTR production |
Approved for ATTR-PN but not for ATTR-CA |
284 mg subcutaneous injection once weekly |
Small prospective imaging-based studies have demonstrated stabilization or improvement in cardiac imaging parameters; a phase II study is currently ongoing |
Thrombocytopaenia, glomerulonephritis, infusion-related reactions and vitamin A deficiency |
|
Eplontersen48,50 |
ASO inhibitor of TTR production |
Approved for ATTR-PN but not for ATTR-CA |
45 mg subcutaneous injection monthly |
CARDIO-TTRansform is a phase III RCT designed to assess the efficacy of eplontersen in patients with ATTR-CA |
Headache and vitamin A deficiency |
|
NTLA-200157 |
CRISPR-Cas9 editing of the TTR gene |
Under investigation |
0.1 or 0.3 mg/kg single intravenous infusion |
A small phase I trial has demonstrated that NTLA-2001 is safe and results in a sustained TTR knockdown |
Headache and vitamin A deficiency |
|
NI00671 |
Recombinant human anti-ATTR monoclonal IgG1 antibody that binds ATTR-amyloid |
Under investigation |
Four-weekly intravenous infusions with doses ranging from 0.3 to 60 mg/kg |
A small phase I trial has demonstrated that NI006 is safe and at higher doses appeared to reduce NT-proBNP, troponin I and myocardial extracellular volume; a phase III trial will assess the efficacy in ATTR-CA |
Arthralgia and thrombocytopaenia |
|
PX00474,75 |
Humanized IgG antibody that binds ATTR-amyloid |
Under investigation |
Four-weekly intravenous infusion with doses ranging from 0.1 to 3.0 mg/kg |
A small phase I trial has demonstrated that PX004 is safe |
Unknown |
|
AT-0276,78 |
Pan-amyloid removal technology using a single-chain fusion protein to stimulate immunological removal of amyloid |
Under investigation |
Intravenous infusion (dosing not available) |
A small phase I trial is currently ongoing |
Unknown |
ASO = antisense oligonucleotide; ATTR-CA = transthyretin cardiac amyloidosis; ATTR-PN = transthyretin amyloid polyneuropathy; CRISPR = Clustered Regularly Interspaced Short Palindromic Repeats; FDA = Food and Drug Administration; IgG = immunoglobulin G;KCCQ = Kansas City Cardiomyopathy Questionnaire; mRNA = messenger RNA; 6MWT = 6-minute walk test; NSAID = non-steroidal anti-inflammatory drug; NT-proBNP = N-terminal pro–B-type natriuretic peptide; RCT = randomized controlled trial; siRNA = small-interfering RNA; TTR = transthyretin.
Figure 1: Disease-modifying therapies for transthyretin cardiac amyloidosis
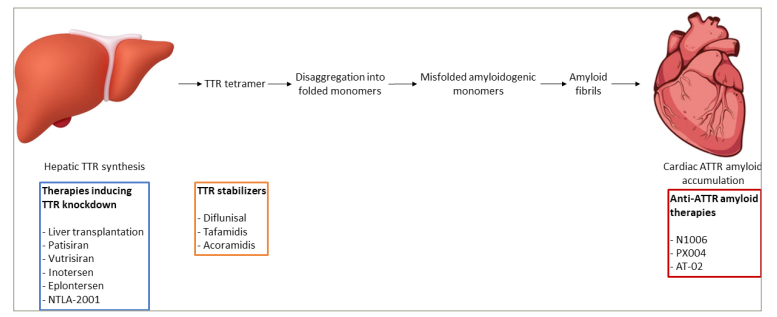
ATTR = transthyretin amyloid; TTR = transthyretin
Conclusion
Advances in the understanding of the underlying pathogenesis responsible for ATTR amyloid fibril formation and the subsequent accumulation in the myocardium have led to the development of multiple novel therapies with different mechanisms of action. Tafamidis remains the only approved therapy for ATTR-CA, but with multiple different agents at various stages of development, and this is likely to change in the near future. The arrival of novel therapies will represent a significant step forward in the treatment of this inexorably progressive and fatal cardiomyopathy.
Despite demonstrating efficacy, many patients still experience disease progression while on treatment with tafamidis. The potential expansion of the armamentarium of disease-modifying treatments will hopefully provide alternative options for those who do not have an adequate treatment response to tafamidis, and hence, there is an increasing need to identify markers of worsening disease that may highlight the need to switch to alternative agents. NT-proBNP progression, outpatient diuretic intensification and worsening 6-minute walk test distance have all recently emerged as markers of disease progression.88,89Alongside imaging-based biomarkers that provide a surrogate measure of the change in amyloid burden in response to treatment, these markers could easily be used in clinical practice to detect patients who may require more intensive therapy.74,75
The development of anti-amyloid therapies represents an important unmet need, and although still at early investigational stages, if successful, these treatments will augment immune-mediated amyloid fibril removal, disease regression and reversal of cardiac disease.
Lastly, considering that, in the contemporary era, patients are being diagnosed earlier in the course of their disease, further research is needed to guide when treatment should be initiated and which treatments should be initiated at each disease stage.9